

Authors
|
|||||||
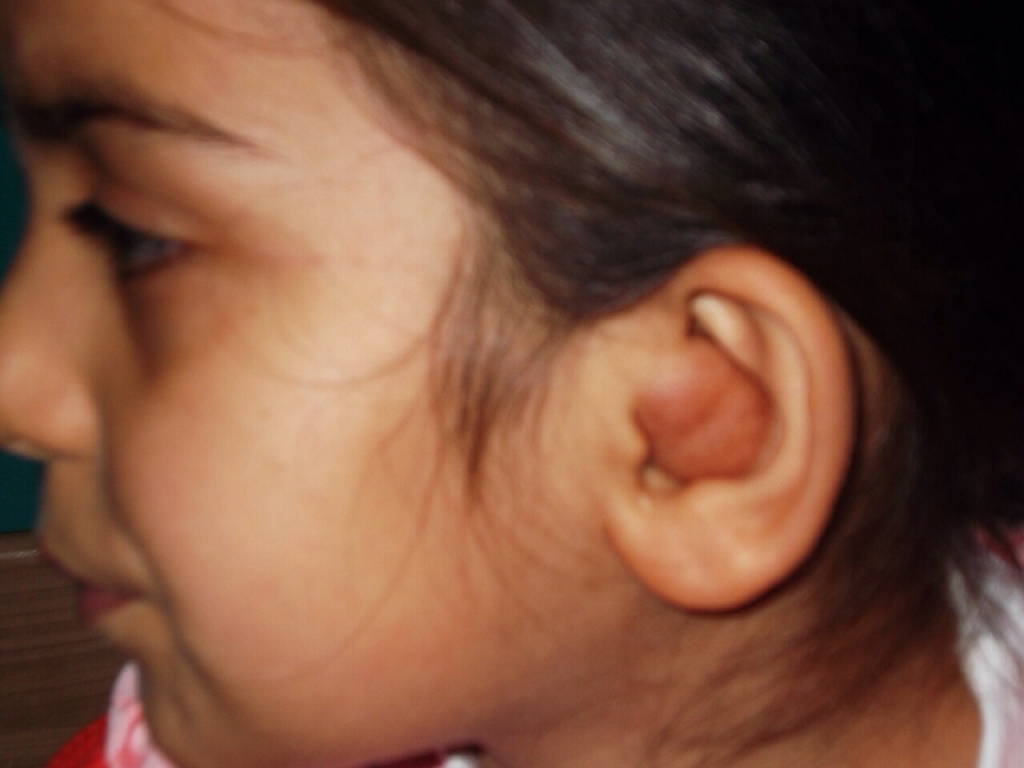
AbstractNeurofibromas are relatively common tumours of the nervous system, but only a few cases involving the external ear have been reported. We are reporting here a case of a 7-year-old girl with neurofibroma of the external ear with no history of neurofibromatosis . The primary complaint was conductive hearing loss and cosmetic deformity. There was complete occlusion of the external auditory canal. The swelling was excised. Surgery resulted in an excellent functional and cosmetic outcome. The aim of this study is to analyze the clinical features of isolated neurofibroma of the external ear canal unassociated with neurofibromatosis type 1(NF1) in the light of the literature.IntroductionNeurofibromas are benign peripheral nevre sheath tumor composed of a variable mixture of Schwann’s, perineurial cells, and fibroblastic cells. They are circumscribed but nonencapsulated neoplasms of nervous system. They can grow anywhere in the body, includes nerves just under the surface of the skin, as well as nerves deeper within the body, spinal cord, or brain. There are many histological subtypes of neurofibroma including localized, plexiform and diffuse [1,2]. Plexiform neurofibroma involves multipl fascicles of a nevre and consists of a proliferation of cells that extend along the length of the nevre. Plexiform neurofibromas are the least common variant considered pathognomonic of the NF-1 [3-5]. External ear involvement by a solitary and peripheral plexiform neurofibroma in patients with no other sign of neurofibromatosis is uncommon. Case ReportA 7-year-old girl attended our clinic with a soft swelling at the left concha, extending into cartilaginous part of external auditory canal and filled the meatus (Figure1).
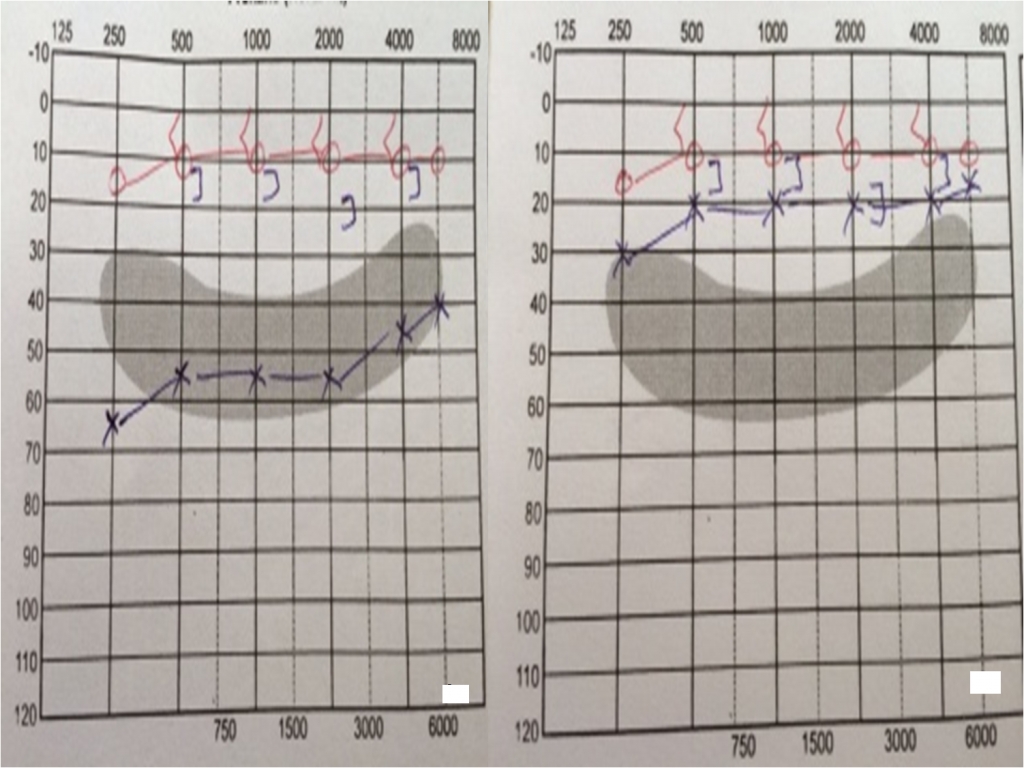
The bony part of the external canal and the tympanic membrane were normal. The size of the swelling was about 4x2 cm. There was no pain and history of bleeding. Pure tone audiometry revealed a conductive hearing loss in the left ear that averaged approximately 55 dB (Figure2).
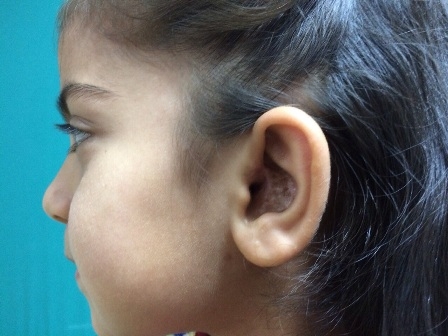
The patient noticed the swelling for the first time at 4 years ago, when it was small. An incisional biopsy was done in our department. The histopathology report was neurofibroma. She had no family history of neurofibromatosis and other lesions of neurofibromatosis, including cafe au lait spots, cutaneous tumors, axillary freckling, Lisch nodules of the iris, optic glioma and bony abnormalities. It was the first case in her family. The swelling was excised along with conchal and external meatal skin under general anaesthesia; the conchal cartilage and perichondrium were preserved. A split-skin graft from the supraclavicular region was used to cover the defect. Histologic examination of the specimen identified it to be plexiform neurofibroma. No significant cellular atypia was seen. The patient made an uneventful recovery, and a good cosmetic result was achieved (Figure3). The conductive hearing loss in the left ear resolved. During 2 year follow-up period there has been no recurrence.
DiscussionNeurofibromatosis (NF) is one of the most common inherited disorders. There are two types of NF, type 1 (NF-1) and type 2 (NF-2). NF-1 affects approximately 1 in 2190 to 7800 individuals. The genetic transmission of NF-1 is characterized by an autosomal dominant pattern, with complete penetrance and variable expression. It linked on a large gene on chromosome band 17q11.2 [6,7]. In 1987, the National Institutes of Health (NIH) developed diagnostic criteria for NF-1. The NIH criteria are a useful guide in a clinical diagnosis of NF-1. An individual is diagnosed with NF-1 if two or more the features identified by the NIH Consensus Development Conference are present [7-9]. A few neurofibromas alone do not indicate NF-1. The present case did not have any other features of NF-1. We have been able to find only one published case about neurofibroma unassociated with NF-1 in the external ear [1]. Approximately, 25% of all neurofibromas are found in the head and neck region [3]. The real frequency of isolated neurofibromas in the external ear is uncertain. Neurofibromas are benign peripheral nevre sheath tumor composed of a variable mixture of Schwann’s, perineurial cells, and fibroblastic cells. They are circumscribed but nonencapsulated neoplasms of nervous system [3]. They can grow anywhere in the body, includes nerves just under the surface of the skin, as well as nerves deeper within the body, spinal cord, or brain. In NF-1 neurofibromas most commonly grow on the skin. The neurofibroma occurs as isolated or multipl lesions frequently associated with NF-1. The clinical behavior of isolated neurofibromas in the head and neck region and the absence of NF-1 association reinforce that sporadic lesion could be hyperplastic or hamartomatous rather than neoplastic in nature [3, 7]. There are many histological subtypes of neurofibroma including localized, plexiform and diffuse [1,2]. Plexiform neurofibroma involves multipl fascicles of a nevre and consists of a proliferation of cells that extend along the length of the nevre. They contain a heterogenous mix of Schwann cells, fibroblasts, and other type cells. Plexiform neurofibromas are commonly found in the head and neck region because of the rich innervation of the area. The trigeminal nevre is the most commonly affect cranial nevre [7]. The plexiform neurofibroma, also named plexiform neuroma, pachydermatocele or neurofibromatous elephantiasis, has been classified as a benign tumor of peripheral nevre. It is highly vascularized, slow growing and locally invasive non-metastatic tumor [9]. Several textbooks have claimed that a plexiform neurofibroma is pathognomonic for NF-1 [10,11]. Plexiform neurofibromas are the least common variant considered pathognomonic of the NF-1 [3-5]. Lin et al reported that ‘ Although plexiform neurofibromas are highly suggestive of NF-1, they are not pathognomonic as claimed [7]. When associated with NF-1, plexiform variant is commonly disfiguring, causing cosmetic abnormalities, pain, functional deficits, and neurologic manifestations with a significant rate of malignant transformation [3-5,9,12]. Solitary lesions are not usually associated with the systemic manifestations unlike multipl lesions which are commonly seen in patients with NF. The frequency of malignant transformation that occurs in solitary lesions unassociated with the disorder is not known but some authors [3,8,13] have reported that NF-1 associated plexiform neurofibromas seem to have an increased incidence of malignant change. There is an approximately %2 to %5 risk of a malignancy [7,9,14]. In spite of plexiform neurofibromas presenting a benign clinical behavior, they are poorly circumscribed, diffused enlargement of neural sheaths that can become distorted into convoluted masses. Dysfunctions including cosmetics abnormalities, pain, and neurologic deficits can be caused by the lesions [3,12]. The exact pathogenesis of the neurofibromas, particularly the plexiform variant, is not completely established. Further studies are necessary to confirm the genetic alterations in sporadic neurofibromas to clarify the exact nature of these lesions in the head and neck region. Theraphy of neurofibromas are controversial. The clinical behavior of neurofibromas is characterized by a benign course with a low frequency of recurrence after surgical excision, but some cause local destruction secondary to pressure effects. [3]. Large lesions located in critical areas, generally associated with NF-1, have a high recurrence rate [1,3]. Trevisani et al, reported that ‘complete surgical excision of these lesions is virtually impossible; if not contraindicated [15]. Needle et al, reported that ‘Tumor progression is a serious problem for children with plexiform neurofibroma [5]. According to Marocchio et al, the clinical behavior of neurofibromas is characterized by a benign course with a low frequency of recurrence after surgical excision [3]. In our case we excised mass along with conchal and external meatal skin. The patient made an uneventful recovery, and a good cosmetic result was achieved. During 2 year follow-up period there has been no recurrence. External ear involvement by a solitary and peripheral plexiform neurofibroma in patients with no other sign of neurofibromatosis is uncommon. Our case showed that plexiform neurofibrom could ocur in the external ear as an isolated lesion, not associated with the NF-1, presenting benign clinical behavior. It seems to have a hyperplastic hamartomatous nature whose pathogenesis needs to be further investigated. References
|
|||||||
| Keywords : Dış kulak kanalı , nörofibrom , İletim tipi işitme kaybı | |||||||
|